The main use-case of Twigstats is to compute f2-statistics for pairs of populations.
This should be straight forward: We compute these using a single
function call in R, using the output files of Relate as input. The
output can then directly be fed into functions of the admixtools2 R
package (https://uqrmaie1.github.io/admixtools/index.html) to
compute any other f-statistic.
Each function implemented in the Twigstats package is described on this page: leospeidel.com/twigstats/reference.
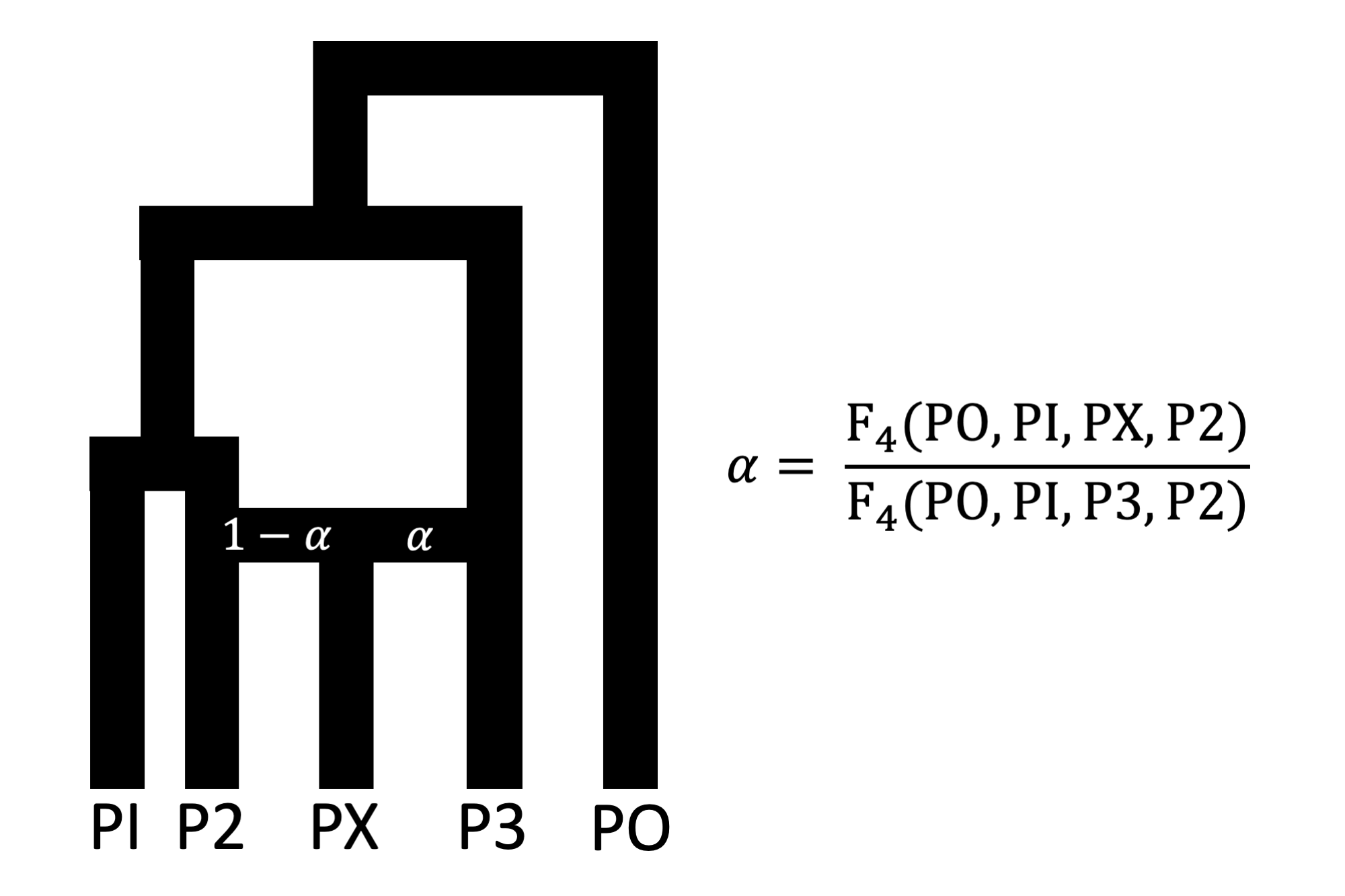
Here, we will demonstrate key features using an example stored under
inst/sim/, which contains simulated data for five
populations as described in our paper (Figure 1).
library(twigstats)
#> Loading required package: dplyr
#>
#> Attaching package: 'dplyr'
#> The following objects are masked from 'package:stats':
#>
#> filter, lag
#> The following objects are masked from 'package:base':
#>
#> intersect, setdiff, setequal, union
#> Loading required package: tidyrComputing f2-statistics from Relate genealogies
The first use-case is to compute f2-statistics on Relate genealogies
(https://myersgroup.github.io/relate/).
The central function is called f2_blocks_from_Relate()
which takes Relate output files as input.
Please type
?f2_blocks_from_Relate for more details:
We will illustrate a basic Twigstats run using example files that are included in the Twigstats package. In this example, population PX is admixed between populations P2 and P3, with a mixture proportion of 20% and 80%, respectively.
These lines assign filenames to variables file_anc,
file_mut, poplabels, and
file_map.
#see https://myersgroup.github.io/relate/getting_started.html#Output for file formats
file_anc <- system.file("sim/msprime_ad0.8_split250_1_chr1.anc.gz", package = "twigstats")
file_mut <- system.file("sim/msprime_ad0.8_split250_1_chr1.mut.gz", package = "twigstats")
#see https://myersgroup.github.io/relate/input_data.html for file formats
poplabels <- system.file("sim/msprime_ad0.8_split250_1.poplabels", package = "twigstats")
file_map <- system.file("sim/genetic_map_combined_b37_chr1.txt.gz", package = "twigstats") #recombination map (three column format)We will first calculate f2 statistics as normal without a
cutoff.
We will then plug these f2 statistics into an f4-ratio
statistic which computes an estimate of the admixture proportion.
#Calculate regular f2s between all pairs of populations
f2_blocks1 <- f2_blocks_from_Relate(file_anc, file_mut, poplabels, file_map)
f4_ratio(f2_blocks1, popX="PX", popI="P1", pop1="P2", pop2="P3", popO="P4")
#> popO popI pop1 pop2 popX val se
#> 1 P4 P1 P2 P3 PX 0.9974217 0.3076228As we can see, the inferred admixture proportion is not close to the true value of 80% and the standard errors are large. This shows that we are underpowered to infer the admixture proportions using this f4-ratio statistic.
Now we will specify a twigstats cutoff of 500 generations and plug these into an f4-ratio statistic:
#Now calculate f2s using a twigstats cutoff of 500 generations.
#This should give us a big power boost.
f2_blocks2 <- f2_blocks_from_Relate(file_anc, file_mut, poplabels, file_map, t = 500)
f4_ratio(f2_blocks2, popX="PX", popI="P1", pop1="P2", pop2="P3", popO="P4")
#> popO popI pop1 pop2 popX val se
#> 1 P4 P1 P2 P3 PX 0.8110266 0.02085804The standard errors are much reduced!
- Use argument use_muts to compute f2-statistics on the (age-ascertained) mutations, instead of branch lengths of Relate genealogies.
- Use argument blgsize to change jackknife block size
- Set argument transitions to False to exclude transitions
Using admixtools2
We can now directly feed this into admixtools2 functions:
library(admixtools)
#no twigstats ascertainment
f4(f2_blocks1, "P4", "P3", "P2", "PX")
#> # A tibble: 1 × 8
#> pop1 pop2 pop3 pop4 est se z p
#> <chr> <chr> <chr> <chr> <dbl> <dbl> <dbl> <dbl>
#> 1 P4 P3 P2 PX 0.00127 0.000314 4.04 0.0000543
#with twigstats ascertainment
f4(f2_blocks2, "P4", "P3", "P2", "PX")
#> # A tibble: 1 × 8
#> pop1 pop2 pop3 pop4 est se z p
#> <chr> <chr> <chr> <chr> <dbl> <dbl> <dbl> <dbl>
#> 1 P4 P3 P2 PX 0.000635 0.0000253 25.1 1.37e-138If PX exclusively clustered with P2 and did not carry gene flow from P3, we expect this f4 statistic to not be significantly deviating from 0. We observe a highly significant attraction of P3 to PX, especially when ascertaining coalescences of the last 500 generations using Twigstats, consistent with PX being a mixture of P2 and P3.
Computing f2-statistics on age-ascertained SNPs
Sometimes, we are prefer not to graft samples into genealogies
(e.g. due to low sequencing coverage, or if we don’t trust the
imputation).
Therefore, the second use-case of Twigstats is to
compute f2-statistics on age ascertained mutations.
The central function is called
f2_blocks_from_RelateAges() which takes plink (bed/bim/fam)
files and mutation ages (.mut) as input.
Please type
?f2_blocks_from_RelateAges for more details:
We will begin with assigning filenames to the following variables. In
our case, all files are stored under path and have prefix
pref.
path <- paste0(system.file("sim/", package = "twigstats"),"/")
pref <- "msprime_ad0.8_split250_1"
file_plink <- paste0(path,pref) #only need prefix
file_mut <- paste0(path,pref) #only need prefix (here same name as plink file but can be different)Next, we compute f2-statistics as normal on the plink format and plug these into an f4-ratio statistic to estimate the admixture proportion.
#Compute regular f2 statistics between all pairs of populations. You can use pops to only calculate f2s between specified populations.
f2_blocks1 <- f2_blocks_from_RelateAges(pref = file_plink, file_mut)
f4_ratio(f2_blocks1, popX="PX", popI="P1", pop1="P2", pop2="P3", popO="P4")
#> popO popI pop1 pop2 popX val se
#> 1 P4 P1 P2 P3 PX 1.082681 0.2931867As before, we can see that the standard error of the inferred dmixture proportion is very large. This shows we are lacking power using the standard f-statistics approach.
We can now ascertain mutations by age. Here we choose a cutoff of 500 generations:
#Use a cutoff of 500 generations
f2_blocks2 <- f2_blocks_from_RelateAges(pref = file_plink, file_mut, t = 500)
f4_ratio(f2_blocks2, popX="PX", popI="P1", pop1="P2", pop2="P3", popO="P4")
#> popO popI pop1 pop2 popX val se
#> 1 P4 P1 P2 P3 PX 0.810077 0.03097378The standard errors are much reduced! So while we are unable to get a reliable estimate without twigstats ascertainment, we estimate PX to carry approximately 80% P2 and 20% P3 ancestry with twigstats.
- Use argument pops to analyse a subset of populations and fam to change population assignment of individuals
- Use argument blgsize to change jackknife block size
- Set argument transitions to False to exclude transitions
- Use file_mut = “./1240k/1240k” to calculate f2 statistics at 1240k sites
- Use file_mut = “./1000G_ages/1000GP_Phase3_mask_prene_nosingle_chr1” to calculate f2 statistics at 1240k sites
Using admixtools2
We can now directly feed this into admixtools2 functions:
library(admixtools)
#no twigstats ascertainment
f4(f2_blocks1, "P4", "P3", "P2", "PX")
#> # A tibble: 1 × 8
#> pop1 pop2 pop3 pop4 est se z p
#> <chr> <chr> <chr> <chr> <dbl> <dbl> <dbl> <dbl>
#> 1 P4 P3 P2 PX 0.00116 0.000305 3.81 0.000139
#with twigstats ascertainment
f4(f2_blocks2, "P4", "P3", "P2", "PX")
#> # A tibble: 1 × 8
#> pop1 pop2 pop3 pop4 est se z p
#> <chr> <chr> <chr> <chr> <dbl> <dbl> <dbl> <dbl>
#> 1 P4 P3 P2 PX 0.000642 0.0000281 22.9 9.35e-116As before, we find a highly significant attraction of P3 to PX using Twigstats, consistent with PX being a mixture of P2 and P3.